Featured publications
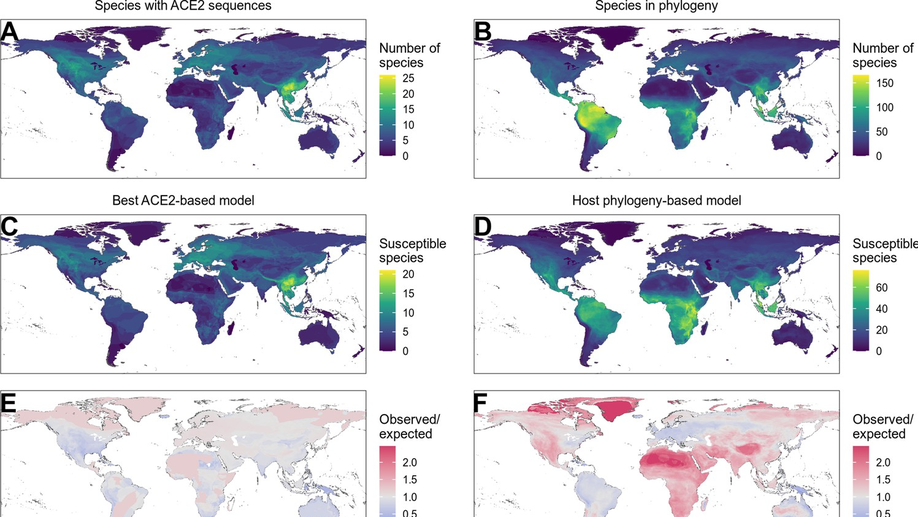
Variation in the ACE2 receptor has limited utility for SARS-CoV-2 host prediction
A large number of publications have shown suprisingly accurate predictions of which species are susceptible to SARS-CoV-2 infection using just the variation in ACE2 proteins found in different species. This would seem to imply that receptor-binding is the primary barrier to cross-species transmission for this virus. However, we show that the predictive power of ACE2-based models derives from strong correlations with host phylogeny rather than processes which can be mechanistically linked to infection biology. Further, biased availability of ACE2 sequences leads to misleading projections of the number and geographic distribution of at-risk species. Models based on host phylogeny reduce this bias, but identify a very large number of susceptible species, implying that model predictions must be combined with local knowledge of exposure risk to practically guide surveillance.
Identifying and prioritizing potential human-infecting viruses from their genome sequences
We describe a machine learning model that identifies candidate zoonoses using evolutionary signals of host range encoded in viral genomes. This allows identification of high-risk viruses immediately upon discovery, increasing both the feasibility and likelihood of downstream virological and ecological characterization and allowing for evidence-driven virus surveillance.
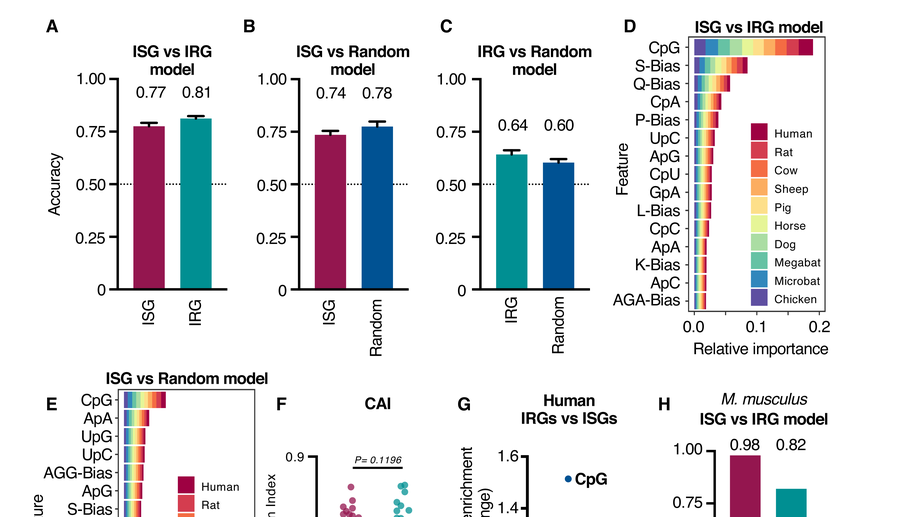
The antiviral state has shaped the CpG composition of the vertebrate interferome to avoid self-targeting
How do vertebrates evolve sequence-specific antiviral defences without accidentally targeting their own gene transcripts? We show that self-targeting by antiviral effectors has shaped the composition of host transcripts in the vertebrate interferome. These unique compositional signatures give us a better picture of what viral genomes capable of evading sequence-specific host defences might look like, an observation we are exploiting in our work to develop genome-based zoonotic risk prediction methods.
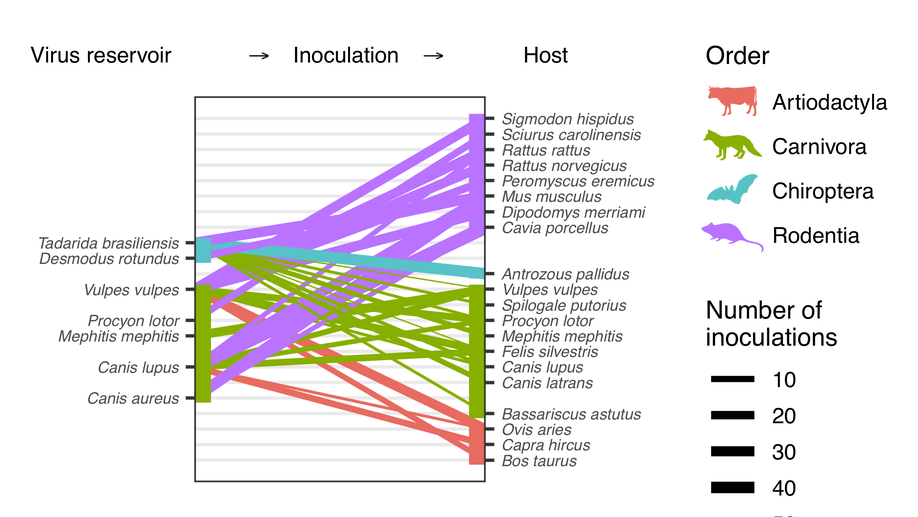
Virulence mismatches in index hosts shape the outcomes of cross-species transmission
Why most cross-species transmissions fail to establish ongoing transmission in the newly infected species remains poorly understood. Examining cross-species inoculations involving rabies, we show that mismatches in virulence which are predictable from host and viral factors make sustained transmission in the novel host less likely. These mechanistic insights help to explain and predict host shift events and highlight meta-analyses of existing experimental inoculation data as a powerful and generalisable approach for understanding the dynamics of index infections in novel species.
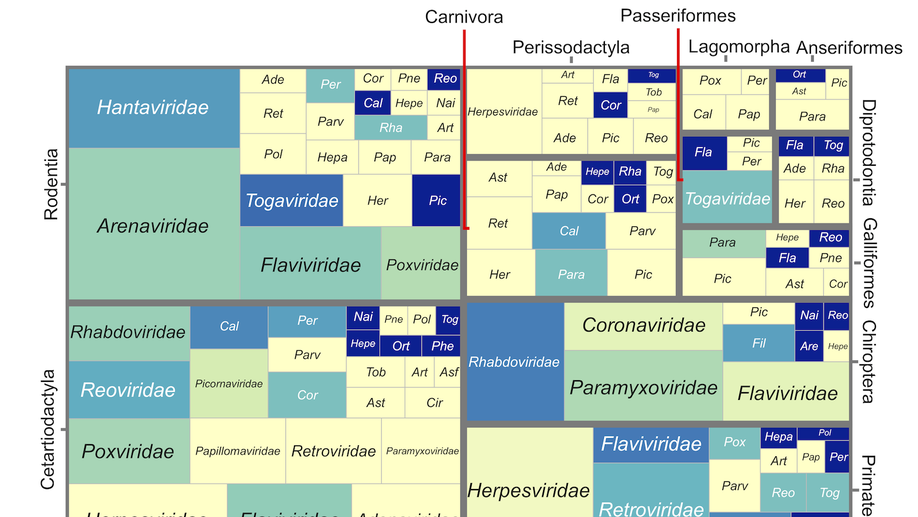
Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts
Do some reservoir groups (e.g. bats) produce more zoonotic viruses than others? By cataloguing the accepted reservoirs for 415 viruses associated with mammals and birds, we show that there is currently no evidence for the existence of any such special reservoir groups. Instead, groups containing more reservoir species are associated with more viruses, and proportionally more zoonotic viruses.